Oxygen Binding Proteins II: Hemoglobin
Myoglobin is admirably suited for storing oxygen and delivering it to the muscle tissue as it is used up. Transport of O2 from the lungs to the tissues through the circulatory system, however, places different requirements on the transport molecules.
O2 transport in vertebrates is carried out by a protein called hempglobin. Hemoglobins also occur in a diverse range of other organisms including some mollusks, nematodes, bacteria and, interestingly enough, some nitrogen-fixing plants. The old jokes about getting blood from a
turnip really weren’t that far off.
Mammalian hemoglobins are heterotetramers consisting of two a and two b chains. Both the a and b chains are very similar to myoglobin in primary, secondary and tertiary structure, and both contain heme bound in the same fashion as in myoglobin.
The human a chain contains 141 amino acid residues; the b chain contains 146. The heme iron is bound to histidine F8 as in myoglobin. In the a chain, His87 is residue F8; in the b chain it is His92. The two a chains are 180o apart, as are the b chains.
 As a result of this (noncovalent) contact
between subunits, any conformational change in one subunit can be transmitted
through the contacts to one or more other subunits. This is the basis of a phenomenon known as cooperative
binding or cooperativity.
As a result of this (noncovalent) contact
between subunits, any conformational change in one subunit can be transmitted
through the contacts to one or more other subunits. This is the basis of a phenomenon known as cooperative
binding or cooperativity.
Cooperativity occurs when binding of substrate by one subunit effects substrate binding by the other subunits – i.e. alters their affinity for substrate. Let’s look at what happens in O2 binding by hemoglobin when compared to myoglobin.
Myoglobin has a p50 of about 4 mm Hg. The pO2 in the lungs is about 100 mm Hg; the oxygen concentration in the peripheral blood vessels corresponds to a pO2 of about 30 mm Hg. Myoglobin is essentially saturated at both these levels; if we tried to use myoglobin as an oxygen transporter, the oxygen bound in the lungs would never be released until the peripheral blood oxygen concentration fell to very low levels. The myoglobin in the muscles would never be fully oxygenated and the muscle tissue would be subject to continual oxygen depletion.
Well, we might suggest, all we need is a myoglobin molecule with a higher p50 to deliver it’s oxygen to the tissues at a higher pO2. This won’t give us what we need, however. The graph shows the hyperbolic oxygen binding curves for myoglobin-like molecules with different p50’s of 4, 30 and 50 mm Hg. Even if we raise the p50 to 50 mm, the protein is still 38% saturated at the venous pO2. Worse, in order to get that degree of dissociation at 30 mm pO2, we have reduced the maximum saturation in the lungs to 67%. In other words, the molecule does a bad job of picking up oxygen where it is available, and an even worse job of releasing it where it is needed.

 What we need is a molecule that is highly
efficient a binding oxygen in the lungs and equally efficient at releasing it
in the tissues. This is what we have in
the cooperative-binding hemoglobin tetramer.
Instead of the hyperbolic binding curve we see for myoblobin, hemoglobin
has a sigmoidal binding curve. The
actual affinity of the molecule for oxygen decreases as pO2
decreases.
What we need is a molecule that is highly
efficient a binding oxygen in the lungs and equally efficient at releasing it
in the tissues. This is what we have in
the cooperative-binding hemoglobin tetramer.
Instead of the hyperbolic binding curve we see for myoblobin, hemoglobin
has a sigmoidal binding curve. The
actual affinity of the molecule for oxygen decreases as pO2
decreases.
 This sigmoidal binding curve is typical of
cooperative proteins, and what it implies is a protein that changes it’s
ability to bind substrate on the fly. At
low concentrations of oxygen, where only a few hemes are occupied, the protein
exists in a low-affinity Tense or T State. At higher pO2 where more and more hemes are
bound by oxygen, the protein enters a high-affinity
Relaxed or R State. The
conversion from one state to the other is governed by the change in
conformation of a bound subunit being transmitted through the intersubunit
contacts subunits to the unbound subunits, pushing them into a state more likely
to bind oxygen. The illustration below
shows how the sigmoidal binding curve can be thought of as the product of two
separate binding curves. At low pO2, the
molecule is in the T state shown by the shallow hyperbolic binding curve. At high pO2, the molecule exists in the R
state and reflects the steep high-affinity binding curve. Transition from T to R results in the S
shaped curve.
This sigmoidal binding curve is typical of
cooperative proteins, and what it implies is a protein that changes it’s
ability to bind substrate on the fly. At
low concentrations of oxygen, where only a few hemes are occupied, the protein
exists in a low-affinity Tense or T State. At higher pO2 where more and more hemes are
bound by oxygen, the protein enters a high-affinity
Relaxed or R State. The
conversion from one state to the other is governed by the change in
conformation of a bound subunit being transmitted through the intersubunit
contacts subunits to the unbound subunits, pushing them into a state more likely
to bind oxygen. The illustration below
shows how the sigmoidal binding curve can be thought of as the product of two
separate binding curves. At low pO2, the
molecule is in the T state shown by the shallow hyperbolic binding curve. At high pO2, the molecule exists in the R
state and reflects the steep high-affinity binding curve. Transition from T to R results in the S
shaped curve.
What Happens in the Protein
In the deoxygenated form of the protein, the iron (and the attached heme ring) are puckered towards Histidine F8. Histidine F8 is, obviously, an integral part of helix F. When O2 occupies the binding pocket and binds the heme Fe+2, it pulls the iron back into the plane of the heme. Histidine F8 is pulled along with it, and this shifts the entire F helix of which the histidine is a part.
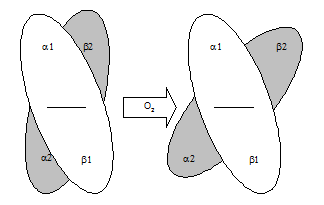 This shift in the F helix has effects on the
interface between subunits, causing movement between the a and b
subunits. In the tetramer, there is
little direct contact between the two a subunits, and little
contact between the two b subunits. The
change in conformation that occurs during oxygen binding primarily involves the
contact between a1
and b2
and between a2
and b1. The net effect is that the a1b1
subunit pair slides over the a2b2 pair, rotating by about 15o.
This shift in the F helix has effects on the
interface between subunits, causing movement between the a and b
subunits. In the tetramer, there is
little direct contact between the two a subunits, and little
contact between the two b subunits. The
change in conformation that occurs during oxygen binding primarily involves the
contact between a1
and b2
and between a2
and b1. The net effect is that the a1b1
subunit pair slides over the a2b2 pair, rotating by about 15o.
In the doxygenated form of the molecule, a number of salt links exist which bind the ends of the polypeptide chains into a fairly rigid arrangement. These links essentially lock the molecule into the low affinity state, and are why the state is referred to as the Tense state. When the heme is pulled flat, the movement of the F helix is transmitted to the adjoining parts of the molecule. The salt links are broken and the constraints locking the molecule in the T state are removed. This shifts the equilibrium between the T and R states and facilitates O2 binding.
Measuring Cooperativity: The Hill Number
Let’s reconsider the hyperbolic binding we saw for myoglobin. We defined an equation for the fractional binding of oxygen, Q, as follows:
![]() (1)
(1)
We can also define a quantity 1- Q as follows:
![]() (2)
(2)
or
![]() (3)
(3)
taking the quotient between (1) and (3), we get:
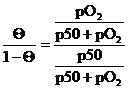 (4)
(4)
Simplifying:
![]() (5)
(5)
If we take the log of both sides of equation 4, we get the following equation:
![]() (6)
(6)
This is the Hill Equation. If we plot log(Q/1-Q) versus log(pO2), we get a line with a slope equal to 1 and an X intercept at the point where pO2 is equal to p50. But lets look at what happens if we derive the Hill equation for a protein with multiple (“n” = some integer) binding sites. We can call it Fooglobin or Fb for short. Furthermore, we assume that this protein binds substrate all at once – i.e. it binds n O2 and fills all it’s binding sites in one fell swoop. We could write the reaction for that protein as follows:
Fb + nO2 ⇌ Fb·nO2 (7)
Then the equilibrium constant will be defined as follows:
![]() (8)
(8)
and the fractional binding equation becomes:
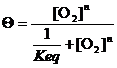 (9)
(9)
or
![]() (10)
(10)
Note: this gives us a sigmoidal binding curve! The Hill equation for this protein then becomes:
![]() (11)
(11)
The slope of this line, n, is equal to the number of binding sites. Real cooperative proteins like hemoglobin deviate from this ideal Fooglobin, however. At very low pO2, real hemoglobin is in the T state; T state binding resembles hyperbolic myoglobin with a high p50, and the Hill Plot at very low pO2 has a slope of 1. At very high pO2, real hemoglobin is almost entirely in the R state. R state binding resembles hyperbolic myoglobin with a low p50, and the Hill Plot at very low pO2 has a slope of 1. At moderate pO2, the transition between T and R states, the Hill plot is steep, and the maximum slope is defined as the Hill Number (nH), which is a measure of the cooperativity of the protein. The minimum value for the Hill Number, as in myoglobin, is 1; the maximum value for the Hill Number is equal to the number of binding sites on the protein – 4 in the case of mammalian hemoglobin. Real human hemoglobin has a Hill Number of around 3.1 – a highly cooperative protein. If one extrapolates the T-state part of the curve, where slope = 1 at low pO2, to the x axis, the intercept is a measure of p50 for the T (low-affinity) state of the protein. Extrapolating the R-state part of the curve to the x axis gives an estimate of p50 for the high-affinity R state.
Other Factors Effecting Affinity
The Bohr Effect
The peripheral tissues of the body are not only net consumers of O2 but are also net producers of CO2. When a person exercises, they not only use more oxygen but they also produce greater quantities of CO2. Dissolved CO2 reacts with water to from H2CO3; H2CO3 is a weak acid and increased concentrations decrease pH in the peripheral tissues.
 The O2 affinity of hemoglobin decreases
with decreasing pH. The binding curve
shifts to the right, a phenomenon known as the Bohr effect. Please note - that is Bohr
effect for Christian Bohr, son of the great physicist – turned – biologist, not
Bore effect.
Physiologically, this improves delivery of oxygen to the tissues in
general, since the peripheral pH will typically be lower than lung pH. It also increases delivery above the normal
level during periods of heavy exercise when high levels of CO2 drive
the pH down a little below normal.
The O2 affinity of hemoglobin decreases
with decreasing pH. The binding curve
shifts to the right, a phenomenon known as the Bohr effect. Please note - that is Bohr
effect for Christian Bohr, son of the great physicist – turned – biologist, not
Bore effect.
Physiologically, this improves delivery of oxygen to the tissues in
general, since the peripheral pH will typically be lower than lung pH. It also increases delivery above the normal
level during periods of heavy exercise when high levels of CO2 drive
the pH down a little below normal.
The mechanism of the Bohr effect involves the protonation of the C-terminal Histidine of the b subunits, Hisb146. Protonation of the histidine results in a positive charge on His b146, which can then form a salt link with Aspb94 in the deoxy (T) conformation, stabilizing the T state of the protein. Increased stability of the R state shifts the equilibrium between T and R to the left.
Carbamylation
Amino groups can react reversibly with CO2 to produce carbamates (carbamic acid is H2NCOOH). This primarily involves the N-termini of the hemoglobin chains:
R-NH3+ + CO2 ⇌ R-NH-COO- + H+
There are a few of important results from this:
1) CO2 transport is improved since some CO2 is now being carried back to the lungs directly by hemoglobin
2) The release of H+ decreases pH and increases the Bohr effect
3) Carbamylated N-termini carry a negative charge instead of the positive charge on a free amino group. The negatively charged group is now capable of forming a salt link to the positive charge on arginine a141. This salt link stabilizes the deoxy (T) form of the molecule and favors O2 release.
Once back in the lungs, where [CO2] is low, the equilibrium for the carbamylation reaction shifts to the left, CO2 is released and the ability to form the salt link ceases.
2,3-Bisphosphoglycerate
The central cavity of the hemoglobin tetramer is loaded with
positively charged residues: Lys b82, His b2, His b143, and the NH3+ terminal group
of each b chain. In the T (deoxy) form this central cavity is
quite large. When the molecule pops into
the R (oxy) state, this central cavity shrinks significantly.
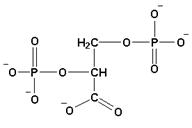 2,3-Bisphosphoglycerate, or BPG (most medical
personnel refer to this molecule as 2,3-diphosphoglcerate) is produced in the
body from one of the intermediates in glucose metabolism,
1,3-bisphosphoglycerate. BPG is highly
negatively charged at physiological pH’s and binds nicely to the positive
charges in the central cavity of deoxyhemoglobin. BPG cannot be sterically accommodated into
the central cavity of oxyhemoglobin, so BPG binding acts to stabilize the T
state of the molecule. As a result,
increases in [BPG] shift the T ⇌
R equilibrium to the left, causing hemoglobin to dump a greater portion of its
oxygen at lower pO2s.
Increases in the concentration of BPG are a primary long-term adaptation
to decreased O2 levels. This can be seen
when people are forced to adapt to high altitudes, and can also be seen in
adaptations to cigarette smoking.
2,3-Bisphosphoglycerate, or BPG (most medical
personnel refer to this molecule as 2,3-diphosphoglcerate) is produced in the
body from one of the intermediates in glucose metabolism,
1,3-bisphosphoglycerate. BPG is highly
negatively charged at physiological pH’s and binds nicely to the positive
charges in the central cavity of deoxyhemoglobin. BPG cannot be sterically accommodated into
the central cavity of oxyhemoglobin, so BPG binding acts to stabilize the T
state of the molecule. As a result,
increases in [BPG] shift the T ⇌
R equilibrium to the left, causing hemoglobin to dump a greater portion of its
oxygen at lower pO2s.
Increases in the concentration of BPG are a primary long-term adaptation
to decreased O2 levels. This can be seen
when people are forced to adapt to high altitudes, and can also be seen in
adaptations to cigarette smoking.
BPG also plays a role in fetal development. Babies in utero are completely dependent on O2 provided by the mother’s bloodstream. Fetal hemoglobin is a teramer of two subunit types as is adult hemoglobin, but different genes are expressed in the child prior to birth. Instead of an a2b2 structure, fetal hemoglobin is contains two a chains and two g chains. In g hemoglobin, His b143 is replaced by a serine residue. This decreases the binding affinity for BPG in the fetal molecule. At any given [BPG], the fetus will always have less BPG bound to hemoglobin than will the mother. At any given pO2, the O2 binding curve for the fetus will always lie to the left of the mother and the fetus will always be able to extract O2 from the mother’s blood. After birth, the g genes shut off and b hemoglobin chains are produced.
Sickle Cell Disease/Trait
Sickle Cell Anemia is a genetic disorder that is characterized by the formation of hard, sticky, sickle-shaped red blood cells, in contrast to the biconcave-shaped red blood cells (RBCs) found in “normal” individuals. This disease is caused by a mutation in hemoglobin; the most common sicke-cell hemoglobin mutation is known as Hb S. Upon deoxygenation, (Hb S) polymerizes and causes red blood cells to change from the usual biconcave disc shape to an irregular sickled shape. The unusual shape of these red blood cells and their propensity to adhere to the walls of blood vessels, can clog the vessels, preventing normal blood flow and decreasing the delivery of oxygen to organs and tissues. These "sickle cells" are also extremely susceptible to hemolysis, causing individuals with sickle cell disease to have chronic anemia.
In addition to chronic anemia, some of the disabling clinical features associated with sickle cell disease include acute chest syndrome, stroke, splenic and renal dysfunction, pain crises in soft tissues and bones and priapism. Children with sickle cell disease are particularly susceptible to bacterial infections.
Sickle cell disease is a major public health concern. This genetic disorder has great impact on both the individual and society. Between 1989 and 1993, there were an average 75,000 hospitalizations per year in individuals with sickle cell disease in the US which, in 1996 dollars, cost $475 million annually.
The gene encoding the beta chain of the hemoglobin molecule, located on chromosome 11, can be mutated in a variety of ways that result in different types of sickle cell disease. Some mutations are more common than others.
Sickle cell disease affects more than 50,000 Americans. Although the disease occurs in high frequency in individuals of Mediterranean, Caribbean, Indian, Arab and Southeast Asian descent, the disease exhibits the highest frequency in people of African descent.
In United States populations, the prevalence of all types of sickle cell disease is equal to 1 in 58,000 Caucasians; 1 in 1,100 Hispanics (eastern states); 1 in 32,000 Hispanics (western states); 1 in 11,500 Asians; and 1 in 2,700 Native Americans, and 1 in 225 in African-Americans.
In addition, 1 in 12 African-Americans are carriers for the disorder (have sickle cell trait). It is believed that the high frequency of the (Hb S) variant is maintained in these populations by the increased resistance to malaria infection in heterozygous carriers, those individuals who possess one copy of the normal beta globin gene (Hb A) and one copy of the sickle variant (Hb S). Individuals who are sickle cell carriers are often referred to as having sickle cell trait, but these individuals do not express symptoms of sickle cell disease.
Hb S involves a change in a single amino acid in the hemoglobin b-chain molecule: Glu6 is replaced by a hydrophobic valine. When these mutant hemoglobins are in the deoxygenated form, the Val at position 6 tends to interact with a hydrophobic pocket on one of the b subunits on a nearby molecule. A valine on that molecule interacts with another, which interacts with another, etc. The net effect is such that the molecules typically group in long bundles of 14 strands, twisted together much like a braid. We can thus readily see the reasons for two aspects of sickle cell disease. First, sickle cell crises (where masses of sickled cells clog the blood vessels clog the capillaries) tend to occur under stress or after heavy exercise when the blood is delivering large amounts of O2 to the tissues and the hemoglobin is largely deoxygenated. Second, sickle cell disease is an autosomal recessive disease – both copies of the gene must be the mutant form for illness to occur. If half the individual’s Hbb is normal, the mutant proteins are unable to form long rod-like polymers.